
Кислота (от латинского корня ac- , «острый»; acere — «быть кислым») — термин, в широком смысле применяемые к свойству любого кислого вещества. В химии наименование кислота имеет более точное значение, обозначая вещество содержащее водород, который может быть замещён с образованием солей. Таким образом, кислоту можно считать солью водорода. Из общих характеристик кислот можно указать, что они растворяют щёлочные вещества и некоторые металлы, нейтрализуют щёлочи и меняют окраску индикаторов синего и фиолетового цвета на красный.
В древности о кислотах было известно не много. Уксус (слабая уксусная кислота с примесями), который получали стоянием вина с доступом воздуха, знали и греки, и римляне, которые считали его типичной кислотой. Это можно даже проиллюстрировать филологически, сравнив слова όξύς, acidus — «кислый» и όξος, acetus — «уксус».
Строение кислот
Другие кислоты стали известны в алхимический период, а первую попытку создать общую концепцию этих веществ предпринял Парацельс, который предположил, что кислоты содержат соединённый «принцип земли и воды». Похожий взгляд высказывал Бехер, назвавший этот принцип acidum primogenium, который, по его мнению, состоял из тех же парацельсовских элементов. Примерно в то же время Бойль исследовал несколько кислот — он установил, что они вызывают покраснение лакмуса, растворяют металлы и щелочные вещества, производят нейтральные тела, или соли, со щелочами. Теоретические концепции взялся вновь рассматривать Шталь, который полагал, что все соли порождаются кислотами, и — ошибочно — что серная кислота является первоосновой всех кислот.
Флогистонная теория окисления и горения привела мысли, что кислоты, получаемые путём обжига, то есть серная, фосфорная, угольная и т. п., нужно рассматривать как элементы. Эта идея господствовала в химии до тех пор, пока не была опровергнута доктриной Лавуазье о том, что кислотообразующим элементом является кислород. Лавуазье пришёл к этому обобщению после опытов со сжиганием неметаллических элементов. Существования кислот, не содержащих кислорода должно было быть достаточно, чтобы доказать несостоятельность этой теории. Но, хотя Бертолле в 1789 показал, сернистый водород (или сероводородная кислота) не содержит кислорода, мнение Лавуазье долго удерживало свои позиции, пока исследования соляной кислоты и хлора Дэви, Гей-Люссака и Тенара, и опыты Гей-Люссака с синильной кислотой не доказали окончательно, что для проявления кислотных свойств наличие кислорода не является обязательным.
В номенклатуре Лавуазье кислоты рассматривались как двойные содержащие кислород вещества, а связанная вода считалась лишь растворителем. Похожие взгляды разделял Берцелиус, который разработал собственную дуалистическую концепцию строения веществ. Позже Берцелиус выдвинул кислородную теорию кислот, тогда как Дэви и, почти одновременно, Дюлонг, стали утверждать, что водород, а не кислород определяет свойства кислот. Оппозиция водородной теории критиковала гипотетические радикалы, которые постулировала кислородная. Кроме того, и электрохимическая теория Берцелиуса скорректировала теорию Дэви и Дюлонга. Согласно системе Берцелиуса сульфат калия представлял собой соединение вида K2O+–SO3−; электролиз должен был разрывать связь между положительным и отрицательным компонентами, после чего окись калия должна двигаться по току, а серная кислота — против него. Эксперименты же показали, что на отрицательном электроде собирается не только калий, но на нём выделяется и водород — это никак не объясняется терией Берцелиуса, зато легко — водородной теорией кислот (калий, образующийся на отрицательном электроде немедленно реагирует с водой, образуя оксид калия и водород).
Дальнейшее развитие теории строения кислот принадлежит Либиху с его концепцией многоосновных кислот. Из идеи Дальтона, что элементы комбинируются преимущественно в эквиатомных соотношениях немедленно последовал вывод, что оксиды, содержащие один атом металла на один атом кислорода образуют одну молекулу нейтральной соли. Эти представления, поддержанные Гей-Люссаком, Леопольдом Гмелиным и принятые Берцелиусом, вели к заключению, что все кислоты должны быть одноосновными. Несправедливость этой теории была доказана исследованиями фосфорной кислоты Томаса Грэма — он показал, что орто-, пиро- и метафосфорная кислоты содержат, соответственно, 3, 2 и 1 молекулы «базовой воды» (которые замещаются оксидами металлов) и одну молекулу фосфорной кислоты, P2O5.
Работы Грэма развил Либих, который исследовал другие органические кислоты — лимонную, винную, циануровую, коменовую и мекониевую — показав, что в этом отношении они напоминают фосфорную. Он также установил как критерий многоосновности кислоты существование сложных солей с различными оксидами металлов. Формулируя эти положения, Либих сначала придерживался дуалистический концепции строения кислот, но вскоре понял, что это не может быть общим правилом, так как галогенные кислоты не содержат кислорода, но образуют соли с такими же свойствами, как и кислородосодержащие, и не вписываются в эту теорию.
Эти и другие причины заставили его отказаться от гипотезы дуалистичности и принятию, как вероятности (и для удобства) положения, что «кислоты являются соединениями водорода, в которых последний может быть замещён металлом», тогда как «нейтральные соли — вещества того же класса, в которых водород замещён эквивалентом металла. Вещества, которые мы именуем ангидридами кислот (оксидами кислот) образуют соли с оксидами металлов, по большей части, при добавлении воды, или разлагающие эти оксиды при высокой температуре».
Водородная теория и доктрина многоосновности выдвинутая Либихом — фундамент современных взглядов на строение кислот. Многоосновная кислота содержит более одного атома водорода, который может быть замещён металлом; более того, возможно и полное замещение, с образованием нормальной соли, частичное — с образованием кислой соли, или замещение атомами разных металлов, с образованием сложных солей. Это можно проиллюстрировать на примере фосфорной кислоты, которая трёхосновна:
| Кислота | H3PO4 | Фосфорная кислота |
| Нормальная соль | Ag3PO4 | Фосфат серебра |
| Кислая соль | Na2HPO4; NaH2PO4 | Кислый фосфорнокислый натрий (дву- и однозамещённый) |
| Сложная соль | Mg(NH4)PO4 | Магний-аммоний фосфат |
| Сложная соль | Na(NH4)HPO4 | Кислый фосфорнокислый натрий-аммоний |
Органические кислоты
Органические кислоты характеризуются наличием в их формуле одновалентной группы –COOH, называемой карбоксильной, в которой атом водорода может быть замещён металлом с образованием солей, и алкильным радикалом с образованием эфиров. Основность органических кислот определяется количеством имеющихся у неё карбоксильных групп.
Оксикислоты — карбоновые кислоты, которые содержат также гидроксильную группу, аналогично мы имеем кетокислоты, альдегидкислоты и т. д.
Классификация. Удобнее всего определить различия между алифатическими и ароматическими кислотами; название первых произошло от названия углеводородов с открытой цепью, а вторых — от кольцевого углеводородного ядра.
Алифатические одноосновные кислоты в свою очередь подразделяются в зависимости от природы родительского угловодорода. Метан и его гомологи дают начала парафиновым кислотам, или кислотам жирного ряда с формулой вида CnH2n+1COOH; этилен — акриловым кислотам вида CnH2n−1COOH, и так далее. Двухосновные кислоты жирного ряда углеводородов имеют общую формулу CnH2(COOH)2n; важными их представителями являются малоновая кислота и янтарная кислота.
Синтез органических кислот. К простым синтезам принадлежат, безусловно, те, при которых карбоксильная группа получается непосредственно от моно- или диоксидов углерода.
Наиболее простые синтезы включают:
- Синтез оксалата натрия пропусканием углекислого газа над металлическим натрием нагретым до 350–360° C;
- Синтез формата калия из влажного углекислого газа и калия, с одновременным получением карбоната калия:
- Синтез пропионата и ацетета калия из углекислого газа, метилнатрия и этилнатрия;
- Синтез ароматических кислот взаимодействием углекислого газа, натрия и замещённых производных брома;
- Синтез ароматических оксикислот взаимодействием углекислого газа и фенолятов натрия (см. Салициловая кислота).
Монооксид углерода (угарный газ) принимает участие в синтезе формата натрия из гидрата натрия, или натронной извести (при 200–220° C); натрия ацетата и пропионтата из метилнатрия и этилнатрия (при 160–200° C).
Другие реакции, которые вводят карбоксильную группу в ароматические:
- Действие хлористого карбонила (фосгена) на ароматические углеводороды в присутствии хлорида алюминия, образующиеся хлорангидриды легко разлагаются водой с образованием кислоты;
- Действие карбамилхлорида Cl–CONH2, циануровой кислоты (CONH)3, выделяющейся синильной кислоты или карбонила на углеводороды при присутствии хлорида алюминия, что приводит к образованию амидов кислоты, легко разлагающихся до кислот.
Важной реакцией синтеза ядра является омыление нитрилов, которые могут быть получены взаимодействием цианида калия и галогензамещёнными производными или сульфокислотой.
Кислоты часто являются продуктами окисления и почти во всех случаях появляются при энергичном окислении. Существуют окислительные реакции, которые могут быть эффективно использованы для синтеза кислота. Первичные спирты и альдегиды, и алифатические, и ароматические, при окислении легко образуют кислоты содержащие то же количество атомов углерода. Ход подобных реакций имеет общий вид:
R–CH2OH → R–CHO → R–COOH–
В случае ароматических альдегидов, кислоты можно получить реакцией Канниццаро (см. Бензальдегид). Важным окислительным синтезом ароматических кислот является их получение из углевородородов с алифатическими боковыми цепями: толуол, или метилбензол даёт бензойную кислоту; ксилол, или диметилбензол — метилбензойную и фталевую кислоты. Кетоны, вторичные и третичные спирты при окислении дают смесь кислот. Также можно отметить разрушение ненасыщенных кислот по двойной связи в смеси двух кислот под действием солей калия.
Выше приведены примеры, при которых карбоксильная группа синтезировалась или вводилась в молекулы. Теперь рассмотрим электросинтез из веществ, в которых карбоксильная группа уже присутствует.
Совершенно очевидно, что металлические соли органических кислот в воде диссоциируют на положительный ион (металл) и отрицательный (кислотный остаток). Эфиры не диссоциируют. Таким образом, смешанная соль и эфир, например KO2C–CH2–CO2–C2H5 даст только два иона, соответственно, калий и остаток молекулы.
Если подвергнуть электролизу раствор ацетата калия продуктами будут этан, углекислый газ, соль калия и водород. Точно так же электролиз раствора янтарнокислого калия даст этилен, углекислый газ, калийную соль и водород, эти реакции можно представить как:
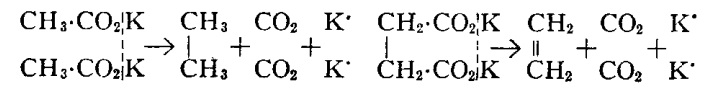
При электролизе раствора этилсукцината калия KO2C–(CH2)2–CO2–C2H5, группы KO2C– отщепляются, а два остатка –(CH2)2–CO2–C2H5 соединяются, образуя эфир (CH2)4–(CO2–C2H5)2.
Тем же путём при электрозе смеси соли металла и эфира могут конденсироваться другие ядра, например, ацетат калия и этилянтарнокислый калий дадут CH3–CH2–CH2–CO2–C2H5.
Реакции. Органические кислоты дают соли металлов с основаниями, и эфирные соли и эфиры вида R–COORˊ со спиртами.
Хлориды фосфора образуют хлорангидриды кислот R–COCl, гидроксильная группа замещается хлором, и ангидриды кислот (R–CO)2O, а молекула воды делится между двумя карбоксильными группами.
Аммонийные соли при нагревании теряют одну молекулу воды и превращаются в амиды кислот R–CONH2, а при дальнейшей дегидратации дают нитрилы R–CN.
Кальциевые соли при перегонке с форматом кальция дают альдегиды; перегонка с натронной известью приводит к образованию кетонов.